Prepared by Dr Korhan OZKAN / Dr. Krishna A. Reddy
Ewing sarcoma (ES) was described by James Ewing. It is a small, round, blue cell neoplasm and is most seen in children and young adults[i]. It has an average incidence of 1.5 per million in adolescents and young adults[ii]. After osteosarcoma, ES are the second most common primary malignant tumors of childhood[iii]. The ES family of tumors includes peripheral primitive, neuroectodermal ES, and extraosseous ES[iv]. ES is pathologically associated with small round cells and t (11;22) (q24;q12) translocation, with FLI1 of the EWS gene or hybrid transcripts of the ERG gene.[4–7] Most cases of ES include chromosomal translocations resulting in t (11;22) (q24;q12) between chromosome 11 and 22. This exchange encodes EWS/FLI fusion protein. The EWS/FLI formation seems to be a critical oncogenic event in the development of ES[v] [vi].
Epidemiology
ES typically occurs in children and adolescents between 10-20 years of age and has a slight male predilection (M:F 1.5:1). 80-85 % of the patients are under 15 years of age and from 20-69% are between 15-19 years of age[vii]. ES predominantly effects Caucasians and rare in Africans, Asians and native Americans[viii].
Location
The most common anatomical sites include the pelvis, axial skeleton and femur. However, ES may occur in almost any bone or soft tissue.
Table 1: Incidence Rates of Primary Sites of Bone Disease[ix]
| Primary Site | Incidence Rate |
| Skull | 5% |
| Spine | 7% |
| Rib | 11% |
| Sternum, scapula, and clavicle | 5% |
| Humerus | 7% |
| Radius, ulna, hand | 2% |
| Pelvis | 18% |
| Femur | 11% |
| Tibia, fibula, patella, foot | 14% |
| Soft tissue | 19% |
Pathophysiology
The call of origin for ES is not fully understood but believed to neuroectodermal in origin. Nonrandom gene rearrangements involving the EWS and the ETS (E26 transformation-specific or E-twenty-six) gene family distinguish Ewing sarcoma. The most frequent gene rearrangement is t(11;22)(q24;q12). A hybrid gene EWS–FLI1 generated by the fusion of the EWS gene on 22q12 with the FLI1 gene on 11q24 occurs in greater than 80% of reported cases. The resulting EWS-FLI1 fusion protein acts as an aberrant transcription factor, which may play a role in the pathogenesis of Ewing sarcoma[x].
The second most common EWS–ETS gene family rearrangement is t(21;22)(q22;q12) translocation, resulting in the fusion of EWS with the ERG gene on 21q22 (observed in about 15% of cases). In the literature, other detected gene rearrangements are t(7;22)(p22;q12), t(17;22)(q21;q12) and (t(2;22)(q33;q12) where EWS is fused with ETS gene family ETV1, E1AF and FEV, respectively[xi]. More chromosomal translocations and complex gene rearrangements have also been reported in the literature. However, whether they are associated with more aggressive tumor features is unclear.
Histology
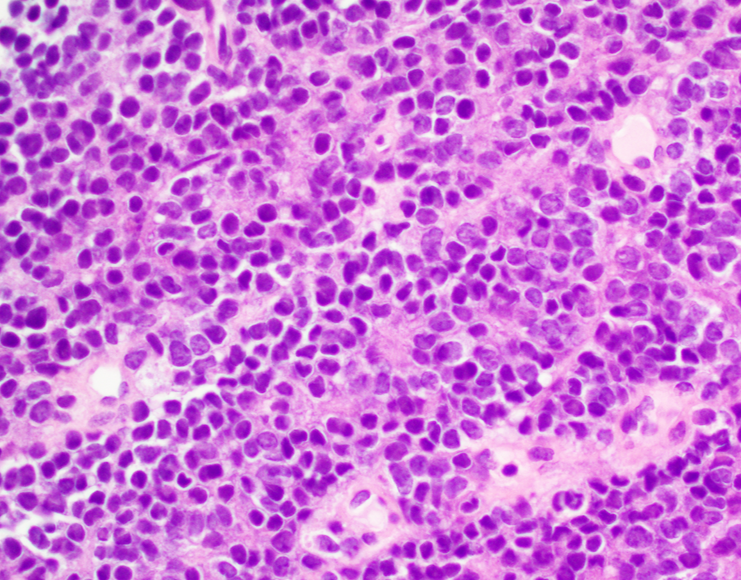
ES is composed of small round cells with an increase in nuclear-cytoplasm ratio that represent family of small round blue cell tumors of childhood (retinoblastoma, neuroblastoma, rhabdomyosarcoma and nephroblastoma). The cells typically stain positive for periodic acid-Schiff (PAS) because of abundant glycogen[xii]. More than 80% of cases exhibit CD99 expression. They also often express CD45, Synaptophysin, chromogranin, vimentin, desmin, S-100 and neuron specific enolase. Molecular genetic studies using immunofluorescence in situ hybridization and reverse transcriptase-polymerase chain reaction are required to make definite diagnosis[xiii].
Imaging Features
Characteristic imaging features include periosteal reaction, bone destruction, and soft tissue mass detected on plain radiographs. Plain radiographs may also show “moth-eaten” lesions, Codman triangle of elevated periosteum or multilayered “onion skin” pattern of periosteal reaction. MRI with and without contrast of the primary site is the choice of investigation. CT thorax, Positron emission tomography (PET/CT), bone scan and MRI pf spine and pelvis can also be used to detect lymph node involvement or metastatic sites[xiv]
Patients who are symptomatic should be referred to the orthopedic surgeon if a biopsy is needed. An open biopsy provides adequate sampling, even though a CT-guided core needle biopsy can establish the diagnosis. The diagnostic workup should include molecular cytogenetic analysis of biopsy specimens to evaluate the t(11;22) translocation.
Bone marrow aspiration with smear and bone marrow biopsy should be considered. The first assessment should consist of serum lactate dehydrogenase, which carries prognostic significance[xv].
Treatment
The standard of care for patients with or without metastasis includes interprofessional treatment with chemotherapy and local therapy, including surgery and radiotherapy. Broadly, systemic therapy is the cornerstone of treatment for all Ewing sarcoma patients. This consists of VDC (vincristine/doxorubicin/cyclophosphamide) with alternating IE (ifosfamide/etoposide). After induction chemotherapy, clinicians typically recommend local therapy using radiation, surgery, or a combination of both. Most institutions use 3-6 cycles of neoadjuvant chemotherapy followed by local therapy and 6-10 cycles of additional adjuvant chemotherapy. For local control, surgery with negative margins is better than radiation treatment alone.
Differential Diagnosis
Other small round cell tumors like neuroblastoma, rhabdomyosarcoma, lymphoma, neuroectodermal tumors, desmoplastic small round cell tumor and synovial sarcoma. Osteomyelitis, osteogenic sarcoma and eosinophilic granuloma should also be considered.
Prognosis
The most important prognostic factor is presence of metastasis at time of diagnosis[xvi]. Localized disease with good response to treatment has approximately 75% five-year survival. Approximately 25% patients with initial localized disease relapse. Survival rates are about 30% in those with isolated lung metastasis and less than 20% in those with bone and bone marrow involvement[xvii].
References
[i] Hassa, Ercan MDa; Aliç, Taner MDb,*. Ewing sarcoma: what trends in recent works? A holistic analysis with global productivity: A cross-sectional study. Medicine 101(46):p e31406, November 18, 2022. | DOI: 10.1097/MD.0000000000031406
[ii] Strauss SJ, Berlanga P, McCabe MG. Emerging therapies in Ewing sarcoma. Curr Opin Oncol. 2024 Jul 1;36(4):297-304. doi: 10.1097/CCO.0000000000001048. Epub 2024 May 22. PMID: 38775200; PMCID: PMC11155282.
[iii] Whelan J, McTiernan A, Cooper N, Wong YK, Francis M, Vernon S, et al. Incidence and survival of malignant bone sarcomas in England 1979–2007. Int J cancer. 2011;000:1–11.
[iv] Singh AK, Srivastava AK, Pal L, et al. Extraosseous primary intracranial Ewing sarcoma/peripheral primitive neuroectodermal tumor: series of seven cases and review of literature. Asian J Neurosurg. 2018;13:288–96.
[v] Delattre O, Zucman J, Melot T, et al. The Ewing family of tumors–a subgroup of small-round-cell tumors defined by specific chimeric transcripts. N Engl J Med. 1994;331:294–9.
[vi] Sorensen PH, Lessnick SL, Lopez-Terrada D, et al. A second Ewing’s sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG. Nat Genet. 1994;6:146–51.
[vii] National Cancer Institute: NCCR*Explorer: An interactive website for NCCR cancer statistics. Bethesda, MD: National Cancer Institute. Available online. Last accessed February 25, 2025
[viii] Jawad MU, Cheung MC, Min ES, et al.: Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome: an analysis of 1631 cases from the SEER database, 1973-2005. Cancer 115 (15): 3526-36, 2009.
[ix] Ewing Sarcoma and undifferentiated small round cell sarcomas of bone and soft tissue (PDQ)- Health professional version (https://www.cancer.gov/types/bone/hp/ewing-treatment-pdq assessed on November 30, 2025)
[x] Lessnick SL, Ladanyi M. Molecular pathogenesis of Ewing sarcoma: new therapeutic and transcriptional targets. Annu Rev Pathol. 2012;7:145-59
[xi] Burchill SA. Ewing’s sarcoma: diagnostic, prognostic, and therapeutic implications of molecular abnormalities. J Clin Pathol. 2003 Feb;56(2):96-102
[xii] Riggi N, Stamenkovic I. The Biology of Ewing sarcoma. Cancer Lett. 2007 Aug 28;254(1):1-10
[xiii] Durer S, Gasalberti DP, Shaikh H. Ewing Sarcoma. [Updated 2024 Jan 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK559183/
[xiv] Ross KA, Smyth NA, Murawski CD, Kennedy JG. The biology of ewing sarcoma. ISRN Oncol. 2013;2013:759725
[xv] Hoshi M, Takami M, Ieguchi M, Aono M, Takada J, Oebisu N, Iwai T, Nakamura H. Fertility following treatment of high-grade malignant bone and soft tissue tumors in young adults. Mol Clin Oncol. 2015 Mar;3(2):367-374
[xvi] Cotterill SJ, Ahrens S, Paulussen M,et al. Prognostic factors in Ewing’s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J Clin Oncol 2000;18:3108-14.
[xvii] Van Mater D, Wagner L. Management of recurrent Ewing sarcoma: challenges and approaches. Onco Targets Ther. 2019;12:2279-2288